Birthdays and Online Conferences go Viral. ISMB2020 & BCC2020 in the time of COVID-19
Meetings In the Time of Coronavirus: Conferences go Viral
June-July usually marks peak conference season when the GigaScience team is on the road, but with the COVID-19 pandemic shutting down travel and public gatherings it has been a strangely static summer. As we launched at the Intelligent Systems for Molecular Biology (ISMB) conference in Long Beach in 2012, it has been a tradition every following year to celebrate our birthday celebrations at the conference. But this year we had to make do with virtual celebrations in the form of a video and #gigascienceat8 social media lookback over our first 8 years.
Conferences have had to go virtual too, and while there were pre-COVID-19 plans to be in Canada attending three conferences, post-COVID-19 we instead have had to participate from the safety and seclusion of our own homes. With different approaches to interaction and tools used to stream these events, they gave us an interesting perspective of how conferences can continue to function and inform in the era of COVID-19. And these conferences had keynotes and sessions directly addressing the COVID-19 pandemic, helping to showcase how these different (but data-centric) scientific fields can quickly pivot and bring their attention onto the biggest issue facing us in 2020.
Montréal was supposed to be the venue for the 28th ISMB 2020 that took place on 13-16 July 2020. As with Medical Imaging with Deep Learning 2020 (MIDL 2020) that was also initially planned to be in Quebec, GigaScience Data Scientist Chris Armit attended this virtual conference in an effort to find out how the Computational Biology community is tackling COVID-19.
The COVID-19 Special Track was a particularly timely and appropriate feature of this conference, and keynote speaker Richard Neher of Basel University set the scene with his talk on “Real-time tracking of SARS-CoV-2 spread and evolution”. As a brief recap, on 31st December 2019, the WHO was informed of cases of pneumonia of unknown aetiology detected in Wuhan, China. By the 3rd of January 2020, a total of 44 patients with pneumonia of unknown aetiology had been reported to WHO by the national authorities in China. On 10th Jan Yong-Zhen Zhang from Fudan University publicly shared the genetic sequence of SARS-CoV-2, the aetiological agent responsible for COVID-19, and it then became possible to understand the genomic properties of the virus responsible for the current global pandemic. A brilliant example of the importance of quickly sharing infectious disease data (see also “the tweenome”), as the roll out of PCR-based diagnostic tests was used for rapid testing and swift release of these data.
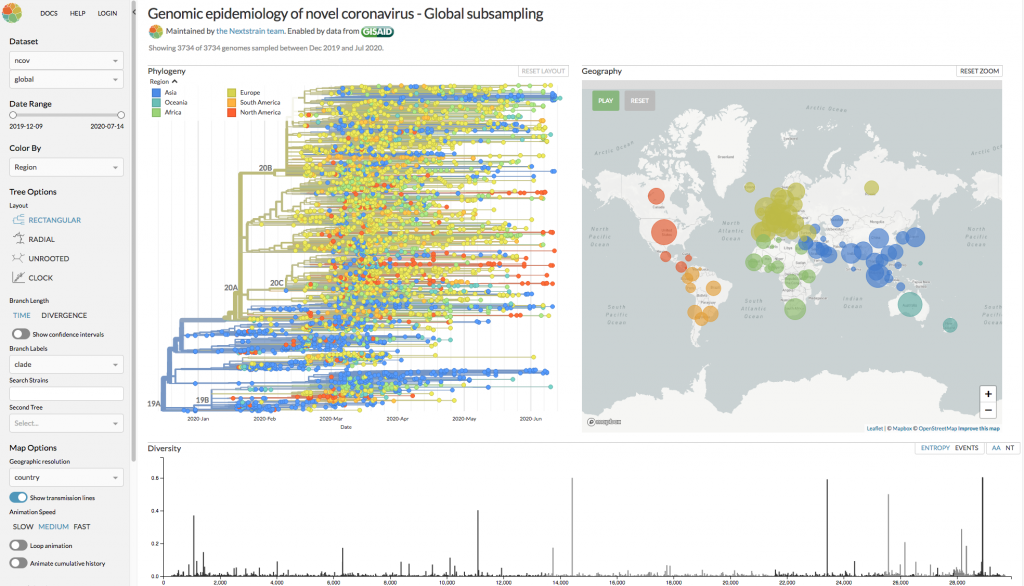
Nextstrain.org allows interactive exploration of genomic epidemiology of novel coronaviruses.
Richard was already exploring evolutionary dynamics prior to the COVID-19 outbreak, and with the open-source project Nextstrain, which focuses on predicting the evolution of pathogens, was able to swiftly identify the relationship between SARS-CoV-2 and other coronaviruses that are found in bats, pangolins, and civets. Of these, one particular strain of coronavirus from bats showed over 96% similarity with SARS-CoV-2, and to date, this is the closest known relative. The SARS-CoV-2 virus has a 29K linear single-strand RNA genome, which is one of the largest RNA viral genomes that have been discovered. Richard explained that by sequence analysis of infected patients, 65,000 complete genomes of the virus are now available for further scrutiny, and this allows us to observe that the virus is evolving. So, a key question is, do the observed mutations in the viral genome correlate with an increase in pathogenicity? Richard was swift to point out that “overall there is no strong signal that the virus has changed in meaningful ways.” However, the mutations are very useful for researchers as they allow us to reconstruct how the virus spreads. Indeed, without sequencing technology, a “transmission tree” of the virus would not be possible. One of Richard’s chief concerns is whether the SARS-CoV-2 spread is seasonal. There is a known seasonality of other viral infections, such as influenza, and this can partially be explained by a greater incidence of indoor rather than outdoor activities in the Winter season. For the transmission of SARS-CoV-2 this is of great concern, as in the Northern hemisphere “the suppression of the curve” has coincided with the arrival of Spring. Richard summed up where we are with the COVID-19 crisis with the rather haunting remark “it is completely in our hands as a society.”
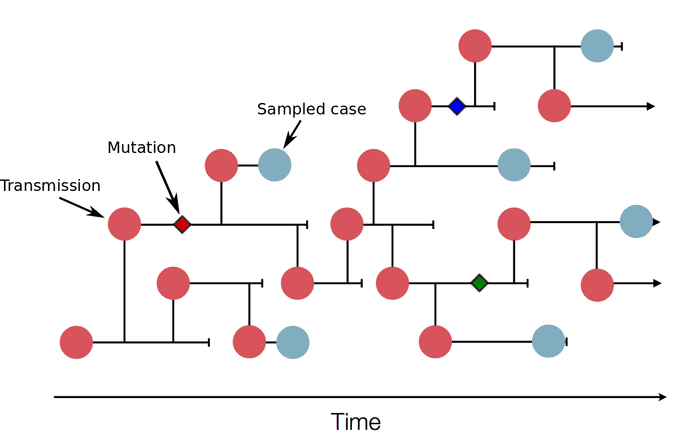
This illustration shows a sketch of a transmission tree with a subset of cases that were sampled (blue). In practice, the transmission tree is unknown and typically only rough estimates of case counts are available. Genome sequences allow us to infer parts of the transmission tree. In this example, three mutations (little diamonds) are indicated on the tree. Sequences that have the same mutations are more closely related, so these mutations allow us to group samples into clusters of closely related viruses that belong to the same transmission chains. Image from: https://nextstrain.org/help/general/how-to-read-a-tree
Despite Nextstrain.org being an open-source project, there was criticism on the openness of the underlying COVID-19 data. GigaScience Editorial Board Member Francis Ouellette of Génome Québec was swift to point out that the GISAID (Global Initiative on Sharing All Influenza Data) Database where the COVID-19 data are archived has restrictive terms and conditions that are “preventing a lot of tool developers from ingesting these data”. At GigaScience, we firmly believe that data and resources should be managed using the FAIR Principles for scientific data management and should be Findable, Accessible, Interoperable and Reusable, and COVID-19 data are no exception in this regard. All our datasets are released under a CC0 public domain to maximise their utility.
So why is SARS-CoV-2 so infectious? A recent publication demonstrated that SARS-CoV-2 entry genes are enriched in nasal goblet cells and ciliated cells within human airways. Specifically, the ACE2 cell surface receptor to which the SARS-CoV-2 spike protein binds, and the protease TMPRSS2 that cleaves the SARS-CoV-2 spike protein and enables the virus to enter the cell, are both enriched in the upper airways. This may explain the highly infectious nature of this pathogen. So is it possible to create a vaccine? Michal Linial of The Hebrew University of Jerusalem followed up on this with her talk on “COVID-19 – The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor”. To gain insight into the high infection rate of the SARS-CoV-2 virus, Michal compared the interaction between the human ACE2 receptor and the SARS-CoV-2 spike protein with that of other pathogenic coronaviruses using molecular dynamics simulations. SARS-CoV, SARS-CoV-2, and HCoV-NL63 recognize ACE2 as the natural receptor but present a distinct binding interface to ACE2 and a different network of residue-residue contacts. A key observation was that the SARS-CoV-2 spike protein has a fastly evolving region that renders the creation of an effective vaccine a significant challenge. Michal further added that cryo-crystallisation experiments and electron microscopy (EM) are key to disentangling this issue.
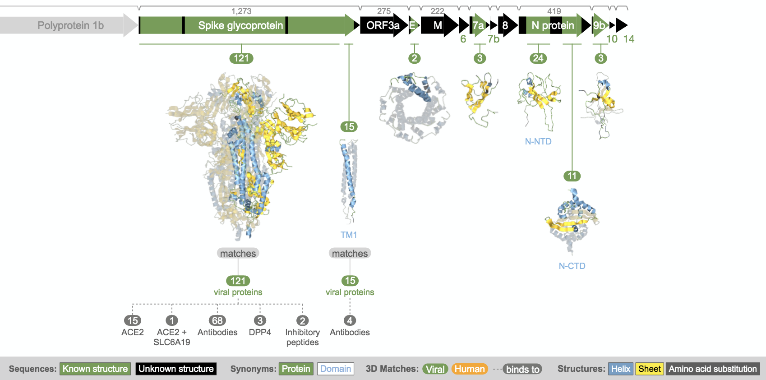
Structural model of spike protein from Aquaria.
So how do we further characterise the SARS-CoV-2 protein structures? Sean O’Donoghue of the Garvan Institute of Medical Research, Australia, gave a very insightful talk on how experimentally determined 3D structures can be augmented by high-throughput generation of homology models, thus allowing researchers to use known structural data to gain detailed insight into the molecular mechanisms underlying COVID-19. The core concept here is that these structural insights will help identify druggable targets for the development of therapies, such as vaccines. I had met with Sean previously at VIZBI 2019 – of which he is the co-organiser – and I was very impressed by his live demos of the protein visualisation tool Aquaria, which has the mission of “simplifying the generation of insight from protein structures” (see GigaBlog of GigaScience at VIZBI). Sean explained that Aquaria now provides ~1,000 3D structure models, derived from all current entries in the Protein Data Bank (PDB), that have detectable sequence similarity to any of the SARS-CoV-2 proteins together with 32,717 sequence features. Sean further pointed out recent enhancements to the Aquaria resource such that it now includes a much richer set of sequence features, including predictions from the PredictProtein and CATH resources.
BioVis and COVID-19
There was an additional focus on COVID-19 at the BioVis track. Hendrik Strobelt of MIT IBM Watson AI Lab delivered his Keynote talk on “Visualization and Human-AI collaboration for biomedical tasks” and showcased his machine-learning model for finding drugs that can treat COVID-19. Hendrik has adapted HITSEE (High-Throughput Screening Exploration Environment), a visualisation tool for the analysis of large chemical screens for the analysis of biochemical processes, to deliver an impressive resource that can enable a user to browse small molecules and to find their closest structural neighbours. The Accelerate Discovery resource utilises a variational auto-encoder, which essentially creates a bottleneck that can be used to learn the latent space of small molecules. This novel AI generative framework has been used to explore three COVID-19 targets and Hendrik and colleagues have thus far have generated 3000 novel molecules.
The Accelerate Discovery resource enables small drug molecules to be screened computationally. Image from: https://covid19-mol.mybluemix.net/
In addition, Josiah Seaman of the Max Planck Institute presented Pantograph, which is a pan genome visualisation tool that captures the genetic diversity of a species in a single graphic. As Josiah explained, graph genomes are an excellent means of visualising genome rearrangements and indels (insertions/deletions), and additionally enable integration with knowledge graphs containing annotations, geographical locations, and patient outcomes. These properties make Pantograph an ideal tool for tracking viral strains of SARS-CoV-2, and without the constraints of a reference genome, viral strains can be smoothly integrated as they are sequenced. To illustrate the power of these visualisations, Josiah showcased a comparison between SARS-CoV-2 and other known SARS viruses.
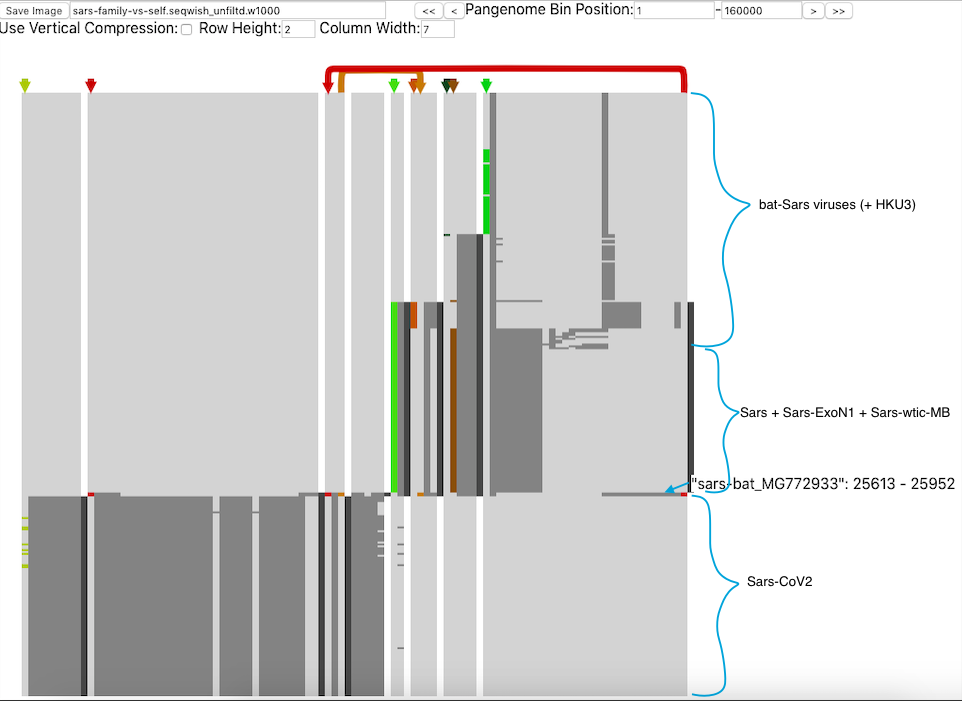
Pantograph comparing different SARS species. Image from: https://graph-genome.github.io/pantograph.html
Additional highlights of BioVis included the talk by Fritz Lekschas of Harvard University on a web-tool called Peax, which enables interactive visual pattern search in epigenomic data using unsupervised deep representation learning. Peax utilises a convolutional autoencoder model to capture visual details of complex sequence patterns. Using this learned representation as features of regions of epigenomic data, Peax then allows users to refine the query by selecting results that they like, and deselecting results that they do not like. This highly intuitive web tool was awarded Best Paper ar EuroVis 2020.
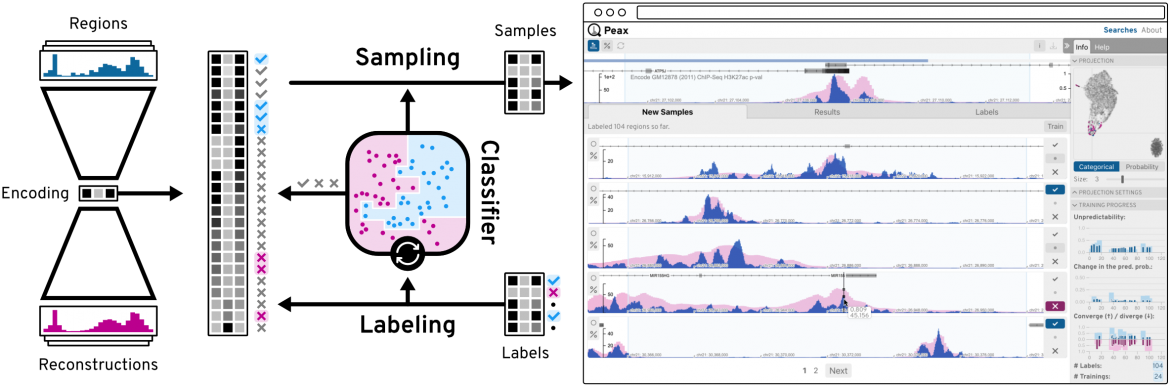
Peax enables interactive visual pattern search queries to be performed on epigenomic data using unsupervised deep representation learning.
Furthermore, there was critical acclaim for Kari Lavikka of the University of Helsinki for the highly impressive GenomeSpy, a declarative, grammar-based approach for specifying interactive genomic visualisations that use the graphics processing unit (GPU). Inspired by “The Grammar of Graphics” – a highly influential book by Leland Wilkinson that detailed a core philosophy on how researchers should approach complex multi-dimensional data – the user-friendly GenomeSpy web interface enables fluid interactions with complex genome visualisations. Kari was awarded the Best Poster Award at BioVis 2020.
Open Source Bioinformatics and Galaxy versus the Coronavirus
The 2nd Bioinformatics Community Conference bringing together the BOSC SIG/COSI historically held at ISMB with the Galaxy Community Conference followed shortly after, and was attended (virtually) by Scott and Laurie from the GigaScience team. We are normally big fans of both meetings and are regular attendees, also linking the Galaxy Conferences to our Galaxy Series in GigaScience. As an Open Science promoting conference, #BCC2020 this year broke down even more barriers, tackling geography and access by presenting all the talks in East and West timezones, and making it much more affordable. This really paid off this year, with over 800 attendees from 62 countries.
As silver sponsors this year, while we didn’t have a physical presence, we had a remote one with Scott and Laurie manning the virtual sponsors table in both time zones and catching up with lots of our old friends, as well as making some new ones. While it wasn’t the same as catching up over birthday cake and drinks, it was still a great experience to interact with familiar faces. And the Remo platform that the BCC2020 organisers used to host the meeting was really excellent for getting across the possibilities of a virtual conference.
Chatting at the @GigaScience table in the poster building at #BCC2020 pic.twitter.com/jOyVA2zUJT
— BOSC (@OBF_BOSC) July 20, 2020
With the concept of virtual conferences going viral, there was no escaping COVID-19 here as well, with our Editorial Board Member Lincoln Stein making his keynote about the advantages and challenges that open source brings to tacking the pandemic. There was a really excellent COVID-19 session that tackled crowdsourced virtual screening of potential COVID-19 drugs, Galaxy workflows for analysing nanopore SARS-Cov-2 data, curated COVID-19 data resources in tranSMART, and a really impressive pipeline (serratus) carrying out a giant cross-SRA search for novel coronavirus sequences, all of the outputs immediately being shared back as CC0 data. Being much more open than the GISAID data mentioned previously.
The one bittersweet thing about the meeting was the fact this was the first Galaxy gathering without Galaxy co-founder James Taylor who tragically passed away in April. There was a lot of talk about JTech – the James Taylor Foundation, which will hopefully carry on much of his legacy in training a new generation of open scientists. We all miss you James.
Pending future COVID-19 waves, next year’s Galaxy Community Conference will be held in Ghent, Belgium on the 5-12 July 2021, and the ISMB Conference is scheduled for 25 – 29 July in Lyon, France. We hope to catch up with many of you there, if not physically then definitely virtually.