DNA day 2020: The genomes of Dominette and Esperanza
It’s DNA day, celebrating two historic milestones: The publication of the structure of DNA in 1953, and the completion of the human genome project in 2003. What better occasion could there be to take a step back and marvel at the breath-taking progress in genomics over the years. For this year’s special DNA day blog, GigaScience authors Benjamin Rosen and Timothy Smith give us insights into the latest in bovid genomics, including a new cattle reference genome and an extremely successful approach to sequence a cattle-yak hybrid with a method called “Trio Binning”. Don’t miss the “Author Q&A” at the end of this article.
We’ve written before on the constant efforts updating and improving agricultural reference genomes with ever improving sequencing technology, and last DNA Day we had a guest post from Sheri Sanders on how to produce an equally high quality genome in non-model organisms. For DNA day 2020, let’s have a look at Dominette and Esperanza.

Dominette
(Fig. from Rosen et al.)
Dominette (or L1 Dominette 01449, as she is known with her full name) was a Hereford cow that gave us the first genome sequence for cattle, published on April 24 2009. Sequencing the bovine genome had high priority at the time, to aid marker assisted breeding and also to understand the domestication history of cattle.
Following in the footsteps of the human genome project, the initial sequencing, assembly and annotation of the first bovine reference genome was a six-year marathon, involving more than 300 scientists who teamed up under the umbrella of the “Bovine Genome Sequencing and Analysis Consortium”. These first assemblies covered approximately 2.5 Gb (=2.5 billion DNA “letters”) and 90% of the total genome sequence could be placed on the 30 cattle chromosomes. At the time, this was a tremendous success. However, 11 years ago, assembling a genome of this size was not an easy task at all, and many gaps remained. Back in 2009, David Burt summarized the state of affairs – and the need to innovate further:
“Genome assembly is still a problem, requiring a combination of parallel computing and hard work from teams of manual annotators, and there is a need for a step change in the algorithms and approaches used to assemble a sequence. “
Fast forward to 2020. Recently, GigaScience published the latest update to the cattle reference genome, using DNA from the same animal that was used 12 years before, Dominette (for more details, also read the author Q&A below).
Since 2009, several technological revolutions took place – notably the invention of “third generation” sequencing, which produces long pieces of DNA information in large quantities, as well as new methods to build bridges over gaps in the genome assembly, to connect loose ends ( so-called “scaffolding”). As the authors point out,
“The development of long-read platforms and a shift from short read to long read sequencing has simultaneously simplified the assembly process and improved continuity and accuracy of the assembly.”
Using a whole range of state-of-the art tools and sophisticated software pipelines, a much smaller team than back in 2009 was now able to produce the best representation of the cattle genome yet:
“The first cattle genome project required a major genome center with large staff, rooms full of instruments, and a budget 50-100 times that of a standard research grant. The assembly we have published now used only a couple of instrument platforms and a budget 400-fold lower, with sequence data collection performed in a single small laboratory.”
The new reference genome has only few remaining gaps, the 30 chromosomes are represented in 345 contigs (continuous segments of sequence).
A lot of the most-important and widely used reference genomes seem to be getting an update, also with a new German Shepherd genome ∼80 times as contiguous as the current canid reference genome just out this month. Likewise, the DNA of the famous TJ Tabasco has enabled a new improved pig reference, which is just about to be published (see the preprint, data in GigaDB, and watch this space for more…).
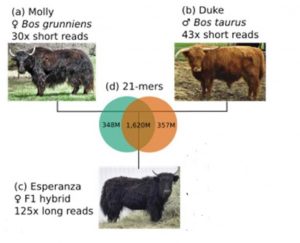
Fig. from Rice et al.
Yattle? Yakow?
So far for Dominette, now let’s talk about another female bovine, Esperanza. She is the main character in another recent GigaScience paper by Rice et al. Rosen and Smith also contributed to this project – again, please do read the Q&A below for more insights directly from the authors.
Esperanza is the daughter of Duke, a cattle bull, and Molly, a Yak cow, and Esperanza’s genome is therefore half cattle, half Yak.
“ The yak is a reasonably close relative to cattle in the phylogeny of species, enough so that interspecies cross can produce viable offspring. The species is adapted to harsh, cold, high-altitude environments and has other phenotypic features that distinguish it from modern domesticated cattle.”
You may call the hybrid offspring a “yattle” or a “yakow”, but in the Himalayas, where the species are cross-bred quite frequently, they are actually called “Dzo”.
The ancient practice of interbreeding Yak and cattle can be traced back to Chinese documents from the eleventh century. Even back then, people knew about a phenomenon that geneticists today call hybrid vigour – essentially, this means that Dzos often produce more milk than either of the parents.
However, For the authors of Rice at al., one main point of sequencing Esperanza was something else than milk or meat: They used the hybrid’s genome and a clever bioinformatic process called Trio Binning to generate genome assemblies of unprecedented continuity.
Our trio binning experiment used an interspecies cross of cattle and yak, with the idea of creating both a yak reference genome and another highly accurate assembly for the Scottish Highland breed of cattle.
In traditional genome sequencing, variability in the genome is a disadvantage. Variable sites are a often causing trouble in conventional genome assembly pipelines and lead to lower-quality results, that is, gappy or inaccurate genome assemblies.
“It worked even better than expected”
However, Trio Binning is a very different game: more variability is actually an advantage for this method – as long as you have access to the DNA of a “trio” of the two genetically diverse parents and their offspring. By producing short pieces of sequence info from the cattle and yak parents, and long reads from Esperanza, their daughter, it is possible to tell for each of the long pieces whether it was inherited from the father or the mother.
“Binning” the sequencing data in this way, it is possible to assemble Esperanza’s Yak and Cattle DNAs separately. Within each “bin” there is no messy ambiguity due to variable sites, therefore it is easier to assemble exceptionally long, continuous stretches of DNA. And the resulting two genome assemblies (one for the Yak, one for the cattle part of Esperanza’s DNA) are “phased” by default, meaning that it is always possible to tell which DNA variants were co-inherited from either Molly or Duke.
Trio-binning as a principle has been performed before, but the new Yak-Cattle has exceptionally long gap-free segments, to the authors’ satisfaction:
It worked even better than expected, with both yak and cattle being the most continuous assemblies of any individual chromosomes of any species, with about a third of the chromosomes being represented by single sequence with no gaps. This was the first time that an assembly had achieved this benchmark without extensive, years-long manual curation and targeted finishing.
Author Q&A with Benjamin Rosen and Timothy Smith
What has changed since the first cattle genome project in terms of technology, but also in terms of the scientific questions?
Rosen & Smith: “Both sequencing and assembly algorithm technologies have vastly improved and reduced in cost. The first cattle genome project required a major genome center with large staff, rooms full of instruments, and a budget 50-100 times that of a standard research grant. The assembly we have published now used only a couple of instrument platforms and a budget 400-fold lower, with sequence data collection performed in a single small laboratory.
The development of long-read platforms and a shift from short read to long read sequencing has simultaneously simplified the assembly process and improved continuity and accuracy of the assembly, greatly reducing the number of gaps and misplaced segments. As a result of these improvements, it is now possible to interrogate more complex regions of the genome that are rich in structural variation and repetitive content such as immune gene clusters. to identify variants affecting health traits. We will also be better able to understand how structural variation contributes to phenotypic variation.
The development of long-read platforms and a shift from short read to long read sequencing has simultaneously simplified the assembly process and improved continuity and accuracy of the assembly. As a result of these improvements, it was possible to resolve complex repeat structures in immune gene clusters to a higher degree, supporting interrogation of these important loci for variants affecting health traits. In addition, there has been a lot of interest in trying to understand how structural variation contributes to phenotypic variation but identification of structural variants can be thwarted by assembly errors. The new assembly should make these analyses more interpretable and repeatable.”
What were particular challenges for the new assembly that is now published in GigaScience?
“One significant challenge was the low heterozygosity (high inbreeding) of the animal chosen for the reference. The technology available for genome assembly 15 years ago was impeded by regions of heterozygosity representing variation between parental alleles. Conventional wisdom of the time was to select the most highly inbred animal available, to reduce the number of regions with high sequence divergence between parental alleles and improve the ability to assemble without confusing segmental duplication with allelic variation.
However, as read lengths kept growing from a few thousand to tens of thousands of bases per read, heterozygosity became a strength by allowing individual reads to be assigned to parental haplotype. Thus, what had been a critical strength of the original assembly with short reads became an impediment to maximizing the quality of a long read assembly of the same animal. Also, detailed physical maps (both radiation hybrid and optical map-based) were available for this animal that required extensive manual curation to determine which dataset provided the best overall fit to all the data in areas where there was disagreement among the assembly and the maps. A high-density genetic linkage map provided final verification of overall structure, but did not have 100% agreement with the assembly produced, again requiring manual curation to determine the most likely “truth” for the genome assembly.”
Why was it important to go back to the same animal that was used in the first cattle genome assembly?
“We placed a high value on removing obstacles towards adoption of the new reference and there were several reasons that this involved using the same animal. The use of the cattle genome is broader than we suspect many readers would realize, as genomics is practiced not just in research but in applied settings in the dairy and beef industries around the world. Millions of animals have had high density genotyping assays performed on them as part of efforts to understand genetic effects on production and health traits, and likely over a hundred thousand cattle have various levels of genome sequence produced. The genotyping platforms and mapping of short read resequencing have all used the original Hereford animal reference.
While we could not prevent the coordinate system from changing because of the many thousands of gaps, misplaced or inverted segments in the first assembly, we believe that use of the same animal minimized the difficulty of “grafting” existing research onto a new reference. In addition, the animal had been sacrificed some years earlier and extensive RNA abundance data had been produced for over 100 tissues derived from her, one of her juvenile calves, and a fetus collected by C-section during mid-gestation. In compliance with the guidelines set up by the Functional Annotation of Animal Genomes (FAANG) international consortium, this data could directly contribute to the effort if we used the same animal for genome assembly.”
What are future challenges? Will there be an even better genome soon?
“One of the major challenges we face is that the reference assembly we are replacing has been in use for many years by hundreds of researchers and commercial operations. The new assembly is orders of magnitude more continuous and many times more accurate by a variety of quality measures, but it is still not perfect. Due to the variety of uses and the intensity of use, those remaining errors are sure to be identified and potentially interfere with targeted research projects, but any correction of such errors then throws off the coordinate system and fixes would require reprocessing of all existing data to fit the new coordinate system.
As mentioned earlier, the use of the highly inbred animal created difficulty in phasing the two parental haplotypes and it is likely there are some genomic areas where contrast in parental alleles has expanded the assembly so that two copies of a genome segment are incorrectly represented where there should be only a single copy, and conversely there are likely areas where there are truly existing tandem duplications on a single parental haplotype that have incorrectly been “collapsed” into a single copy representation. In addition, it has been becoming clear from results in the biomedical research community that a single reference genome is not adequate to represent the full scope of genome variation in a species. For example, some human populations actually have additional genes compared to the human reference, and/or are missing some genes found in the reference. This discovery has led to the notion of developing “pangenome” representations for a species, in which all existing genomic segments are included even though the specific representation does not actually exist anywhere in nature.
Another challenge is how to best utilize multiple reference quality genomes for cattle. In fact, we have just launched a Bovine Pangenome Consortium with the goal of generating over 120 reference quality genomes for cattle and their wild relatives. Thankfully there is a lot of exciting work being done in the area of pangenomes and we are hopeful that by the time our assemblies are generated, the issue of how to represent and manipulate a multiple-assembly-based pangenome will be resolved.
Further improvements in single molecule sequence read quality and length since we generated the data for this assembly, including a completely different platform for long read sequencing that generates even longer reads, and the development of methods for haplotype-resolved assembly called “trio binning” has led to the creation of more highly continuous assemblies for other cattle. However, at present none of those assemblies has applied the full range of data that was available for the Hereford reference, so the intensive manual curation that the Hereford assembly underwent is made much more difficult and to our knowledge has not been performed (and since we were involved with most of them we are pretty sure of that statement).”
Let’s also talk about your paper on the cattle-yak hybrid. In simple words, what is Trio Binning, and how does it help to produce a higher-quality assembly?
“Each of us inherit genetic material from both of our parents, in the form of two complete sets of chromosomes in each cell, one from each parent. During genome assembly, the DNA extracted for sequencing is a mixture of both parental haplotypes. For earlier genome assemblies, the algorithm had to make a choice on what to do when there appeared to be two different forms of essentially the same sequence. Usually, the choice would be to choose one representation over the other at each place along the assembly where this occurred. Since there was no way to assign each choice to the parent of origin, this led to the assembly switching back and forth from one parental allele to the other along each chromosome, making the output assembly a mosaic of the parents. In addition, sometimes the assembly process would instead decide that the two parental alleles were both present on the chromosome and create a false tandem duplication, or conversely where there was true tandem duplication it would collapse the two copies into a single copy based on the mistaken decision that there were just two different parental alleles being observed.
Thus inaccuracies were introduced into the process. In trio binning, we sequence each parent with inexpensive short read sequencing and identify short bits of sequence that are uniquely found in each parent. The long reads produced from the offspring being sequenced for assembly are then individually searched for the presence of these short sequence fragments, which identifies whether the read came from the chromosome inherited from the sire or dam (father or mother).
The reads are then sorted into bins depending on which parent they were derived from. The sire-dam-offspring set of animals is a called a trio, and the process bins the reads prior to assembly, so the procedure is known as trio binning. Then each parental genome can be assembled separately using different parameters that recognize that no decision on parental allele needs to be made so if a segment appears to be present in two copies, it must be a tandem duplication and so a more accurate depiction of the genome can be derived.
Our first trio binning experiment used a cross between two subspecies of cattle, known as Bos taurus taurus and Bos taurus indicus and represented in our study by the Angus and Brahman breeds, respectively. A cross between these two breeds was sequenced and assembled by the trio binning approach, improving in some ways on the Hereford reference assembly including the fact that it was a male so we obtained assembly of the cattle Y chromosome that was not a part of the female Hereford reference assembly. Our second trio binning experiment used an interspecies cross of cattle and yak, with the idea of creating both a yak reference genome and another highly accurate assembly for the Scottish Highland breed of cattle. This worked even better than expected, with both yak and cattle being the most continuous assemblies of any individual chromosomes of any species, with about a third of the chromosomes being represented by single sequence with no gaps. This was the first time that an assembly had achieved this benchmark without extensive, years-long manual curation and targeted finishing.”
Why did you decide to sequence a Yak/cattle hybrid?
“There were technical and biological reasons to sequence a yak/cattle hybrid. The yak is a reasonably close relative to cattle in the phylogeny of species, enough so that interspecies cross can produce viable offspring. The species is adapted to harsh, cold, high-altitude environments and has other phenotypic features that distinguish it from modern domesticated cattle. In addition, both wild and domesticated yak populations exist, presenting opportunity to examine the impact of domestication on the genome. Following the success of the Angus x Brahman trio, we wanted to see if assembly would be further enhanced by increasing the heterozygosity of the offspring even further by interspecies crossing, enabling even more accurate sorting of long reads into the proper parental bins. In addition, the concept of a cattle pangenome was beginning to form and it represented an opportunity to add another breed of cattle with high quality genome assembly. The individual chosen for sequencing was identified living on a farm in Iowa, having been a result of a Scottish Highland bull mating with a yak cow, so we did not have to begin by trying to create such a cross.”
What were particular challenges for this project?
“The first challenge was finding the subject animal. We had help from local yak producers in the USYAKS organization, who successfully located and procured the animal for us. They also bought the yak mother and keep her for breeding with the rest of their herd in Nebraska. There really weren’t any other particular challenges, because the sequencing methods had been well worked out in the previous projects, and the initial assembly came out so amazingly continuous that we were all very pleasantly surprised. We did have a challenge finding a lead person to complete finishing of the genome (which includes assigning and ordering of sequence segments around the relatively few gaps, properly orienting them to established coordinates, error checking and other quality control measures, and assigning everything to specific chromosomes), but we were fortunate to find a very competent postdoctoral associate in a collaborating laboratory, Dr. Ed Rice, to carry the load for us.”
References
Rosen BD, Bickhart DM, Schnabel RD, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience. 2020;9(3):giaa021. https://doi.org/10.1093/gigascience/giaa021
Associated data in GigaDB: http://dx.doi.org/10.5524/100669.
Rice ES, Koren S, Rhie A, et al. Continuous chromosome-scale haplotypes assembled from a single interspecies F1 hybrid of yak and cattle. GigaScience. 2020;9(4):giaa029. https://doi.org/10.1093/gigascience/giaa029
Associated data in GigaDB: http://dx.doi.org/10.5524/100710