Mock the Metagenome. Author Q&A with Nick Loman & Sam Nicholls
The mock metagenome, MAGs and breaking the first rule of Long Read Club

Nick showing us some of his experiments with an early antecedent Short Read Club…
Out today in GigaScience is a new “mock metagenome” Data Note from the Nick Loman lab in Birmingham showcasing the latest long-read sequencing technologies from Oxford Nanopore. Having published the first nanopore E. coli genome with us in 2014 showcasing the then new MinION, this new benchmark presents long read metagenomics datasets from Oxford Nanopore’s new PromethION sequencer. In one of our long running series of author Q&As Nick Loman and first author Sam Nicholls give us the lowdown on the importance of the mock metagenome, open science, MAGs, and how this work is part of a new Long Read Club.
What does a mock metagenomics dataset give you that you can’t get from “real-world” datasets? Why are these community standards important?
Mock communities are really useful for both lab and bioinformatics “positive controls”.
Because the community has gram-negative, gram-positive and two types of yeast cells, it can be used to check that DNA extractions have worked properly. Some kits will fail to lyse some of the cells and they will simply be missing from your experiment! Having a gold standard reference for each genome is really useful for assessing how well bioinformatics aligners and de novo assemblers are working. And the use of known taxa and abundances in invaluable for assessing taxonomic assignment pipelines.
In this case, to keep pace with such changes and to ensure robustness of results, it is incredibly helpful to have validated, known “gold standard” datasets. In this case we used a commercially-available “mock community” from Zymo: real cells, in known abundances, comprised of 10 different microbes.
As genomics is moving into real-world uses such as in the clinic, it is even more vital that experiments are conducted correctly and sequencing and bioinformatics pipelines can be validated. We are trying to help the community design accepted best practices for long read metagenomics.
Our paper is a starting point for a longer-term goal to generate reliable trusted data sets, and make them freely available for the community.
This example presents large amounts of long-read sequencing data, so what does this technology bring to the field of metagenomics?
Metagenomics is a very exciting technique, and the field has really taken off in recent years, and has been used for diverse applications including diagnosis for clinical microbiology, the discovery of new genes and species with potentially industrial biotechnology applications, and for studies of microbial diversity: for example, whole new swathes of the Tree of Life, such as the Candidate Phyla Radiation were discovered using metagenomics!
The field is enthusiastic about being able to recover whole genome information directly from samples without the need for culture, and so-called metagenome-assembed genomes (MAGs) are being collected at an increasing pace. MAGs are usually made by sorting sequence reads or contigs with similar composition or coverage into genome ‘bins’. However, these binned MAGs can be fragmented, miss important genes and end up being contaminated with sequences from related species.
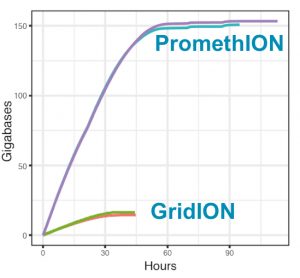
Output of the PromethION & GridION sequencers (source: Nicholls et al.).
In comparison to short-read high-yield platforms where read lengths are limited between 100 and 300 bp, single-molecule long-read platforms can sequence much longer fragments of DNA: with read lengths averaging over 10 Kbp, with over 2 Mbp reported by the community. Long reads simply hold much more information, making them a powerful tool for metagenomics. For example, their increased information content offers more evidence for sequencing mapping against references, or taxonomic databases. Additionally, longer reads are key in bridging repetitive structures within and between genomes.
We think long reads can help with this problem: particularly as the cost of generating long reads has recently dropped dramatically (in our paper we show outputs of ~15Gb from a single MinION flowcell, and ~150Gb from the PromethION flowcells). However, there’s not a clear view on the best way to analyse long read metagenomic data as it’s such a new field.
You gather Oxford Nanopore PromethION, GridION and Illumina data, so what do you see from these comparisons, and what do you hope users will get from this mock metagenome data?
The goal of this work was to make available a really deep long read dataset for the two Zymo mock community standards.
These datasets can be used for a wealth of comparisons: between the evenly-distributed and log-distributed samples, and between the Oxford Nanopore GridION and PromethION platforms too. The Illumina short-read data demonstrates that high-quality short-reads cannot adequately assemble the communities alone, but are useful for polishing our long-read assemblies to a high consensus accuracy (although we found that polishing long-read nanopore data alone with Racon and Medaka does a very good job too).
Our work also benefits from the recent release of PacBio data for some of the isolates in this community from Chris Mason and Alexa McIntyre. These genomes now permit us to make a reliable estimate of the correctness of the assemblies we generate, both in terms of base quality and assembly structure.
We are really hoping that users will take parts (or all!) of our data and use it to develop and validate new laboratory and software pipelines for long-read metagenomics. We’ve taken care to ensure we are up to date with the latest ONT software including the newest “flip-flop model” Guppy basecaller, the quite recent wtdbg2 assembler, and current polishing strategies (Racon, Medaka, Pilon). This is a nice demonstration of what can be achieved with long-read metagenomics, but this is just the beginning and there will be plenty more to come!
The Oxford Nanopore PromethION is a pretty new beast, so what does this platform allow you to do that previous sequencing technologies were not able to do? Especially for the field of metagenomics.
PromethION data is quite sparse at the moment and to our knowledge this is the first metagenomics PromethION dataset to be released. To make a simple analogy, it’s like having a more powerful microscope. Using the PromethION, we see 10-fold increases in coverage for each of the genomes in the community compared to exactly the same library when sequenced on the GridION. This level of resolution permits us to assemble some of the lower abundance organisms from the log-distributed community and to detect organisms at extremely low abundance with more confidence. This will be crucial for some applications where important community members are not the highest abundance in the sample. We also are able to demonstrate that the PromethION data are of similar quality to the GridION when run on the same library, giving confidence that this beast may be useful.
Can you tell us about the Long Read Club, and what does this work contribute to it?
 The first rule of Long Read Club is you don’t talk about Long Read Club…!
The first rule of Long Read Club is you don’t talk about Long Read Club…!
If we were to break that rule, we’d tell you that Long Read Club is part of a recently funded Wellcome Trust Technology Development Award to Nick here in Birmingham, and Matt Loose at the University of Nottingham. The goal is to help the genomics community at large achieve finished-quality complete genomes from long-reads, routinely and at scale. Our work here is a small stepping stone contributing towards the future of long-read metagenomics. We’ve shown that long-read based assemblies are getting close to the single-contig finished quality genomes that one would expect to get from isolate sequencing directly, and we’d like to think that we’re sharing the best extraction, sequencing and bioinformatics protocols currently available.
Most importantly, Long Read Club is a Club, so the core idea is to share our knowledge with each-other and the wider community. Our reads, software and analyses are open access and open source, because we want everybody to be able to achieve really very long reads indeed.
All the data, snakemake workflows and protocols are provided openly without restriction, and your lab have been big advocates of open source approaches to science (e.g., the Tweenome). Have you seen the benefits of this first hand?
We have! The paper itself features recently published data from Alexa McIntyre et al’s recent Nature Communications paper that included PacBio isolate sequencing of eight of the ten organisms in the two ZymoBIOMICS community standards, which was generously shared with us ahead of publication; allowing us to perform robust comparisons of our long-read nanopore assemblies against orthogonal long-read PacBio assemblies. We are very grateful for their early data sharing, not least because it allowed us to respond to a request from the reviewers to include assembly comparisons in this manuscript.
Last month, Mikhail Kolmogorov and Jeffrey Yuan (et al.) published the Flye assembler in Nature Biotechnology. Over the past year, Mikhail and Jeffrey have been developing a metagenomic assembly mode for Flye (‘metaFlye’) – a preprint has just been made available – and have shown it works really well using the Zymo mock community data. We actually made the first version of this dataset available only a few weeks after generating it and released it at last September’s Genome Science meeting. It’s gratifying to know that there are developers taking our data and working to develop and validate new methods for long-read metagenomics. If you’re interested, you can hear more about Flye from Mikhail and Jeffrey in their recent Long Read Club episode.
We want to make everything we do available for everyone to use and improve. The MinION has helped to democratise sequencing and in the past couple of years we’ve seen huge improvement in long-read technology, but it is natural that it takes time for a new field to figure out the best way to analyse data. Making our ideas, protocols and software open means we can accelerate science and work with the genomics community to make long-read metagenomics robust, scalable and scientifically-accurate for everyone.
Further Reading
Nicholls SM, Quick JC, T Shuiquan, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience. 2019 doi:10.1093/gigascience/giz043
Further Watching
Watch the latest updates for the Long Read Club on their youtube channel: https://www.youtube.com/c/longreadclub