Open CycloneSeq Benchmarking For Complete Bacterial Genomes
As another DNA Day treat GigaByte today publishes new benchmark data and analysis of the new CycloneSEQ platform, a novel sequencing technology using nanopores that demonstrates here the ability to sequence complete bacterial genomes.

Getting our hands on a CycloneSeq sequencer
Following on from the recent official launch of BGI’s new CycloneSEQ single-molecule sequencer the new Data Release published today in GigaByte presents the first independently peer-reviewed benchmarking data. The CycloneSEQ platform delivers long-reads using newly developed nanopore technology. This study tests the performance of the new platform in sequencing diverse microbial genomes, and presents both the raw and processed data to enable others to scrutinise and verify the work. To provide further transparency and insight in this benchmarking process, the peer-reviews, raw data, scripts and protocols are also shared for the first time to establish trust in this new technology. By directly reading DNA molecules without fragmentation, the study demonstrating CycloneSEQ delivers long-read data with impressive length and accuracy, unlocking gaps that short-read technologies alone cannot bridge.
Open Science is the way
It is always challenging to convince potential users of the utility of new technologies and present their features and qualities in an unbiased way, but our strict open data and open peer-review policies work well here by throwing light and increasing scrutiny of how these assessments are achieved. GigaScience Press already has a lot of experience showcasing new sequencing technologies in this open manner, publishing the first ONT microbial reference genome in 2014 (the raw data shared in GigaDB helping convince the community that nanopore sequencing was finally feasible), and the same transparent approach was used in 2017 to showcase the first human genome published using BGI’s first DNBSEQ sequencer, the BGISEQ-500. For this new work you can scruitinise the data and peer review reports yourselves from the paper, and one of the reviewers has even blogged their peer review (bringing back #titusischucknorris memories of the real-time blogged reviewing of our Assemblathon 2 paper). Following our recent promotion of the protocols.io platform, protocols have also been cited and embedded in the paper, allowing readers to better understand how the system runs.
The Challenge: Filling the Gaps in Millions of Bacterial Genomes
While long established short-read sequencing technology remains cost-effective and accurate, it struggles to assemble complete, circular bacterial genomes—leaving critical gaps in our understanding of microbial functions. The researchers from BGI-Research have combined CycloneSEQ long-reads with DNBSEQ short-reads (another technology that we published benchmarked data in 2017), achieving closed, circular, high-accuracy genomes for common gut bacteria.
As a standard validation approach, the researchers sequenced the bacterial reference strain Akkermansia muciniphila ATCC BAA-835, yielding 12.07 Gbp of long-reads with an average length of 11.6 kbp. The hybrid assembly method outperformed both short- and long-read-only approaches, successfully assembling a complete genome of ATCC BAA-835 with a mismatch rate lower than 0.0001%. The researchers subsequently sequenced 10 common strains isolated from the human gut, successfully closing all 10 bacterial genomes—plus tiny phage/plasmid circles missed by short reads alone. Using long-read-only assemblies managed to put together complete, circularized genomes for 8 out of 10 strains, while using the previous generation short-read-only assembly failed to produce a single complete genome. The researchers also revealed that the issue is not the inability of short-read sequencing to detect relevant regions but rather the inherent limitations of short-read assembly.
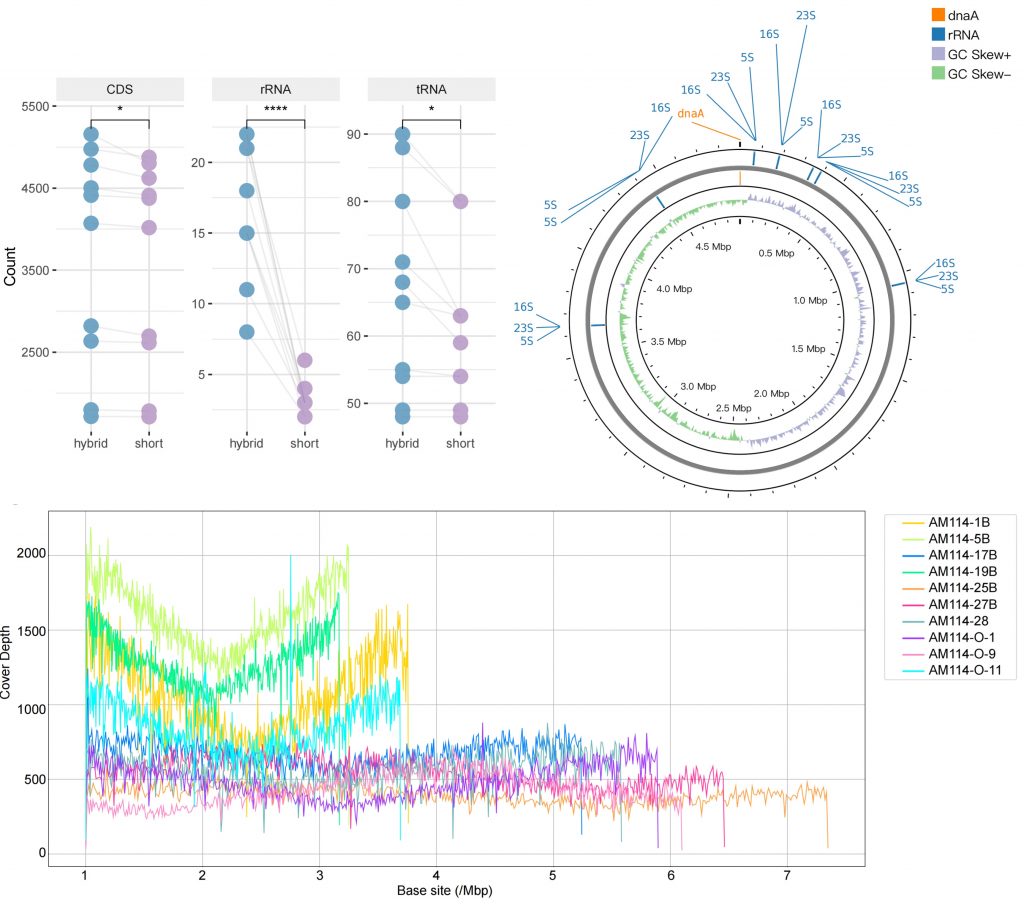
Comparison of complete genomes and short-read assemblies.
In this work the technology also was tested with complex microbial communities. In a 21-strain synthetic gut community (18 bacteria, 2 fungi, 1 archaeon strains), hybrid assembly using CycloneSEQ + DNBSEQ outperformed single-method approaches, yielding 5 complete metagenome-assembled genomes (MAGs)—which was not achieved by short- or long-read assemblies alone.
This new data demonstrates CycloneSEQ’s long-reads already deliver sufficient length and quality for circular genome assembly, but short-reads remain essential for polishing accuracy. The authors’ state that they are now working on expanding this benchmarking to real samples, fine-tuning the balance between short-read and long-read data for even faster, higher-quality assemblies.
References:
Hewei L, et al. Efficiently Constructing Complete Genomes with CycloneSEQ to Fill Gaps in Bacterial Draft Assemblies. GigaByte. 2025. https://doi.org/10.46471/gigabyte.154
Liang H; Zou Y; Wang M; Hu T; Wang H; He W; Ju Y; Guo R; Chen J; Guo F; Zeng T; Dong Y; Zhang Y; Wang B; Liu C; Jin X; Zhang W; Xu X; Xiao L (2025): Supporting data for “Efficiently Constructing Complete Genomes with CycloneSEQ to Fill Gaps in Bacterial Draft Assemblies” GigaScience Database. https://doi.org/10.5524/102694
Hewei L, Wang M. CycloneSEQ Library Construction and Sequencing Protocol for Isolated Bacteria. protocols.io. 2025. https://dx.doi.org/10.17504/protocols.io.kqdg3k3kev25/v1
Quick J, Quinlan AR, Loman NJ. A reference bacterial genome dataset generated on the MinION™ portable single-molecule nanopore sequencer. Gigascience. 2014 Oct 20;3:22. https://doi.org/10.1186/2047-217X-3-22
Huang J, et al. A reference human genome dataset of the BGISEQ-500 sequencer. Gigascience. 2017 May 1;6(5):1-9. https://doi.org/10.1093/gigascience/gix024