Accessing and reviewing Controlled Access Data of Rare Cancers. Q&A with Matthieu Foll
 We have a Q&A with author Matthieu Foll from the WHO International Agency for Research on Cancer (IARC) on his new GigaScience paper presenting multi-omic data from rare lung neuroendocrine neoplasms and his experience having the precious Controlled Access data peer reviewed by named peer reviewers.
We have a Q&A with author Matthieu Foll from the WHO International Agency for Research on Cancer (IARC) on his new GigaScience paper presenting multi-omic data from rare lung neuroendocrine neoplasms and his experience having the precious Controlled Access data peer reviewed by named peer reviewers.
As a journal focused on reproducibility of research GigaScience has a strict open-science policy, meaning access to the data and software supporting our papers needing to be available to peer reviewers and readers. This is usually relatively straightforward with studies on microbes, animals, and plants (even when 13TB in size), but for human research if data is potentially identifiable there are legal and ethical challenges making this more difficult to do. A lot of human sequencing data is now hosted in controlled access repositories such as dbGAP and the EGA, where researchers can gain access the data after applying to a Data Access Committee (DAC) and meeting guidelines relating to the research they wish to carry out. In the past when working with papers containing this type of data we would try to make the brokering process easier for readers by providing the data access forms alongside links to the repository (see this example in GigaDB). To aid reviewers we would usually encourage the authors to provide a representative anonymized dataset, or provide a link to public datasets that could be tested in a similar manner, but this is not the same as having hands-on access to the actual real-world data supporting the results of the study.
Newly published in GigaScience we see an example of the benefits of our mandatory Open Peer Review policy, where a DAC allowed qualified peer reviewers access to inspect the data in their repository as they were named and met the criteria of access (read the actual review here). We’ve written in this blog and in Editorials of the benefits of Open Peer Review and this is another example supporting why we and a growing number of other journals (e.g. PLOS and Nature journals) have moved towards this fairer and more transparent model. To give an author’s insight we provide one of our long running author Q&A articles with lead author Matthieu Foll, who is a Bioinformatics Scientist in the Genetic Cancer Susceptibility Group of IARC in Lyon.
Your paper is a Data Note describing a multi-omic dataset from rare lung neuroendocrine neoplasms, so why did you produce this data?
The molecular characterization of tumors has revolutionized our understanding of cancer biology, how cancer is classified, and how it is treated in the clinic. Large national and international initiatives have performed multi-omics characterization of most common cancers, but several rare cancer types or subtypes have been omitted from these studies, even if they collectively account for ~25–30% of all cancer diagnoses and 25% of cancer deaths. This lack of biological knowledge and consequently, limited therapeutic opportunities translates in a five-year survival on average worse for rare cancers (47%) than common cancers (65%). In this context, we have established the Rare Cancers Genomics initiative (www.rarecancersgenomics.com): an international multidisciplinary open-science effort to shed light on the molecular characteristics of rare cancers, to understand the etiology and carcinogenesis processes, to ultimately improve their clinical management and consequently, their prognosis. In this context we have recently produced the first multi-omics data of the rare pulmonary carcinoids, that was the missing piece for a complete molecular characterization of lung neuroendocrine neoplasms (representing approximately 25% of all lung cancers), which also include the poorly differentiated and highly aggressive lung neuroendocrine carcinomas – i.e., small-cell lung cancer and large-cell neuroendocrine carcinoma. We have integrated this dataset with five previously published datasets to generate the first comprehensive molecular map of lung neuroendocrine neoplasms.
What are the challenges of working with such rare tumours, and how does this rarity effect the value and what others can do with their data?
One of the main limitations we are facing is the lack of available biological material suitable for molecular studies due to the rarity of these diseases. Using fresh frozen tissue samples is preferred for molecular studies because of the higher quality of DNA/RNA they provide, while Formalin-Fixed, Paraffin-Embedded (FFPE) remains the tissue preservation method of choice in clinical settings. However, FFPE tissue processing and storage result in degraded DNA and RNA, limiting gene detection and introducing sequencing artefacts. As a consequence, rare cancer studies are usually based on small series of samples and are therefore underpowered to draw meaningful conclusions. It is therefore of the utmost importance to provide the full spectrum of data and tools to allow other researchers to re-use our data together with a future series of samples.
What issues did you have with sharing this data? As we have named, open peer review, how did you find this process and how did it assist review of the data?
Human genomic data is regarded as personal data and can generally only be shared with controlled-access, where only researchers who apply for and get permission will be able to access it after signing a data access agreement. This is incompatible with anonymous peer-reviewers accessing the data, and the named open peer review policy from GigaScience is therefore a necessity for such datasets. In practice one reviewer requested the data access like other researchers usually do through the EGA platform where our data is hosted, the Data Access Committee granted him access and he was able to review the data.
You also share data, source code, and compute environments via a Nextjournal interactive notebook supplement to the paper, so what advantages does this resource bring, and what do you hope users will do with it?
We wanted to allow anyone to be able to re-use our data, including researchers with limited bioinformatics or biostatistics expertise or infrastructure. Therefore we made a particular effort not to only provide raw sequencing data and final results but also intermediate standardized re-processed as well as reproducible, open-source, easy to use bioinformatics pipelines and source code. We also provide an interactive map hosted on the UCSC TumorMap portal that can be explored like any geographical map simply using a web browser. The entire process to reproduce entire analyses is described in an interactive computational notebook hosted on Nextjournal, and one can easily update the molecular map by integrating new samples starting from raw sequencing reads.
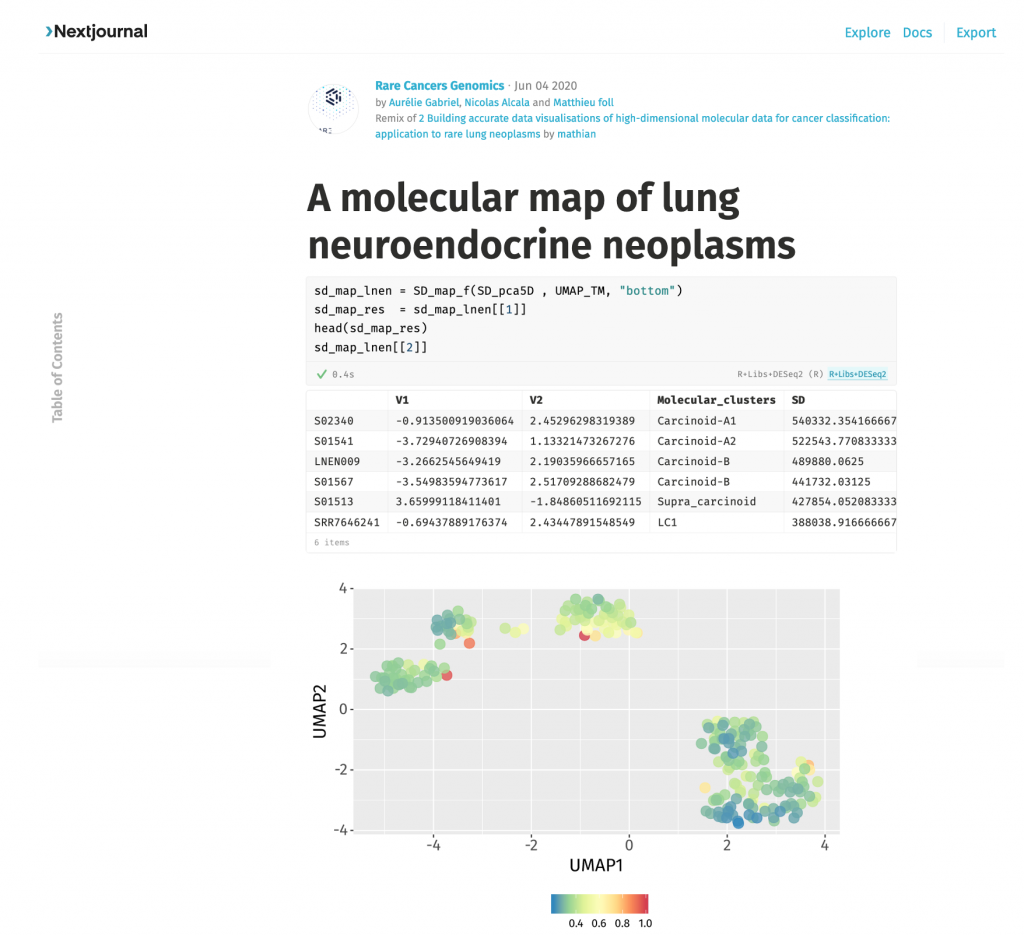
Promoting such data integration will empower more reliable statistical testing, and this map will therefore serve as a reference in future studies, ultimately leading to a better understanding of this rare understudied disease. In the medium term, as high throughput sequencing technologies start to be used for personalized medicine, these molecular maps could be used as a reference map to locate patients, helping for the diagnosis and decision-making.
References
Gabriel AAG, Mathian E, Mangiante L, Voegele C, Cahais V, Ghantous A, McKay JD, Alcala N, Fernandez-Cuesta L, Foll M. A molecular map of lung neuroendocrine neoplasms. Gigascience. 2020 Oct 30;9(11):giaa112. doi: 10.1093/gigascience/giaa112.
Gabriel AAG; Mathian E; Mangiante L; Voegele C; Cahais V; Ghantous A; McKay JD; Alcala N; Fernandez-Cuesta L; Foll M (2020): Supporting data for “A molecular map of lung neuroendocrine neoplasms.” GigaScience Database. http://dx.doi.org/10.5524/100781