DNA Day 2019: How to sequence the genomes of the weird and wonderful
It’s DNA day, commemorating the publication of the structure of DNA in 1953, as well as the completion of the Human Genome Project in 2003. Genomics has come a long way since then. Today it is possible to sequence whole genomes with a very reasonable investment of time and money. What an amazing time for scientists who are working with non-model organisms. As an example, please re-visit our previous DNA Day’s special, an interview with Aaron Pomeranz on #JungleOmics (here). However, the options and parameters to decide upon before embarking on a genome project are bewildering. Where to start? For this year’s DNA day, it is our pleasure to host a guest blog by Sheri Sanders, a bioinformatician at the National Center for Genome Analysis Support (NCGAS). Below she discusses some of the planning steps you need to think about before sequencing your favorite non-model organism. Please also make sure to visit the NCGAS blog and their “Third Generation Sequencing Update” (here), with more in-depth coverage of technical considerations.
A guest blog by Sheri Sanders, NCGAS
The advent of Third Generation Sequencing (TGS) has again changed the landscape of genome assemblies. Third generation sequencing, with its long reads and linked reads for scaffolding, allow reference-quality genomes to be assembled at relatively low costs, opening the door for many non-model genomes to be generated. The success of TGS has fueled projects such as the Vertebrate Genome Project (VGP, website here) and much higher standards for genomes – complete chromosomes and haplotype resolution in critical areas (gold) and even full length haplotype resolution across the entire genome (platinum). TGS has driven this new wave in high quality genomes for non-model organisms, such as those described here, opening up a wide range of research questions, such as the Avian Phylogenomics Project’s full phylogeny of birds found here.

Understanding the evolution of birds with the help of comparative genomics. Figure from “The birds of Genome10K“, courtesy of Rob Davidson.
Third Generation Sequencing is a rapidly changing field and staying on top of the latest practices can be as intimidating as the standards. As a general start point in planning, we at the National Center of Genome Analysis Support (NCGAS) currently suggest the following as a general plan for genome projects on non-model critters (basically the cheaper, non-platinum version of the VGP’s workflow, here):
- High Accuracy Short Reads: 30-50X Illumina
- Less Accurate Long Reads : 30-50X PacBio
- Some Form of Mapping/Scaffolding Technology: Bionano or Hi-C
The approach isn’t anything revolutionary, it is more or less the blueprint of many genome projects these days, especially for non-model organisms or projects with less funding or previous sequencing. The concept is that there is a trade-off between high accuracy sequencing and reads long enough to scaffold your data into contiguous regions through successive approximation.
Bionano provides great long-range architecture information, but by nature of its technology (see here for an overview), only 1Kb resolution. Hi-C technologies (summarized here) also provide this low resolution, structural information. These technologies serve as a very hazy blueprint for genome sequence, but are the main drivers of chromosome-level assemblies. The structure is there, but to get more resolution, more data is needed.
Long-read technologies can then be layered on top of the structural blueprint. PacBio still seems to dominate for general genome sequencing (overview here), but there are several other options–10X (overview here) and Nanopore (overview here) for example. Whatever the technology, long-reads are still a bit less accurate and more expensive than short-reads, but this layer increases the resolution of the map, giving you single-base resolution. However, the accuracy requires consideration.
The most popular way to deal with accuracy issues is to layer on Illumina sequencing, which is still the most accurate and cheapest (though PacBio is catching up!). This gives a high resolution assembly of the genome.
An excellent example of this formula can be found in this paper on the Tegu genome, summarized in this blog from NCGAS and this post in GigaBlog. The VGP adds in 10X sequencing, which is good for haplotype information, but does increase cost.

The genome of the tegu lizard Salvator merianae is the most complete assembly of any reptile genome so far.
One size doesn’t fit all—especially with non-model systems!
While there are two mapping technologies (with their own pros and cons, described here), the most flexible part of this is the sequencing technology. PacBio is increasingly more accurate and getting cheaper, giving Illumina a run for its money. However, each has biases that must be considered. One of the easiest ways to correct for the biases is to use both, but quality genomes have been done with each individually (again, with mapping/scaffolding technologies). An updated review of the decisions involved in using PacBio and Illumina, separately and in conjunction, are reviewed here.
However, one of the fun parts of working with non-model organism systems is that the genomes are unique in some aspect or another. It is critical to consider these genome- and project-specific aspects in your sequence project design. A couple of examples are below and more considerations can be found here.
Genome Structure Considerations
Genome size
The first consideration is genome size. Since most of the recommendations for successful assembly are based on coverage, larger genomes obviously require more sequencing. But the considerations don’t stop there. Larger genomes are large for a reason–polyploidy, repetitive content, and large-scale gene family expansion all contribute to inflated genome sizes. Unfortunately, all make assembly more difficult, even with adequate coverage. In these cases, the repetitive regions must include non-repetitive regions to provide context—meaning longer reads are necessary.
For example, salamanders have particularly difficult genomes to sequence, being an order of magnitude larger than most vertebrates and highly repetitive. The recently successful strategy (details here) was to drastically reduce the Illumina sequencing in favor of more long-read PacBio libraries.
Axolotl Genome

The axolotl: An unusual amphibian that does not undergo metamorphosis. Photo: LoKiLeCh , CC-BY-SA, via Wikipedia.
{kind=link}
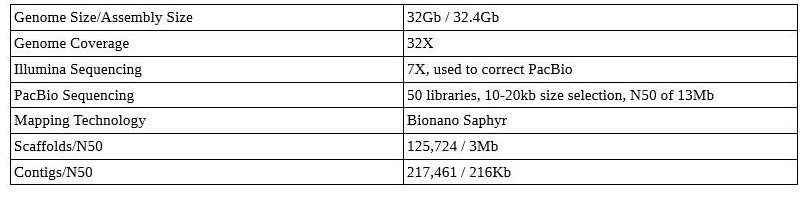
Notice those contig/scaffold counts—this is not a chromosome-level assembly! Even with TGS, large genomes can be difficult and may not be as well assembled as you’d expect with similar sequencing on a smaller genome.

Non-model, non-flowering plants a. Picea abies b. Ginkgo biloba c. Cibotium barometz d. Adiantum caudatum e. Marsilea crenata f. Asplenium viride g. Diphasiastrum complanatum h. Bryum capilare i. Marchantia polymorpha Fig 6 from “10KP: A phylodiverse genome sequencing plan” (individual photo credits are here) .
{kind=link}
Heterozygosity
Heterozygosity should also be an early consideration in planning your genome project. One of the largest contributors to mis-assembly/fragmentation in genome assemblies, especially when using PacBio data, is haplotype variability. Haplotypes can differ structurally or at the sequence level, and lead to breaks in the continuity of the assembly.
There are a few ways to deal with this issue. The first is nothing new—inbreed your organism to remove heterozygosity. This option is still very valid and very common. However, it can be difficult in non-model organisms with longer generation times. If you cannot inbreed, another way to get low heterozygosity is to aim for haploid genomes–via tissue selection (i.e. gametes), sequencing technology (10X shines here), or experimental design. The third option includes trio binning, which has become a hot topic.
Trio binning (open access pre-print here) is a relatively new experimental design to make high-quality genomes from diploid organisms. Instead of trying to create low heterozygosity individuals, trio binning leverages existing heterozygosity to isolate parent genomes in F1 hybrids. Light sequencing (30X Illumina) is required of both parents, which is then used to “bin” deeper coverage of the F1 (~40X PacBio) into reads that are from each parent genome. Since the F1 contains a haploid genome of each parent, you can assemble each “bin” separately and end up with high-quality haploid genomes for one both parents species/breeds/individuals at once.
While this design does increase the required Illumina sequencing (30X for each parent, deeper coverage of the F1), two genomes are produced and mapping technology is not necessarily required (though I’d still recommend budgeting for at least Bionano).
Examples of Trio Binned Genome Sets (from the Nature paper here)
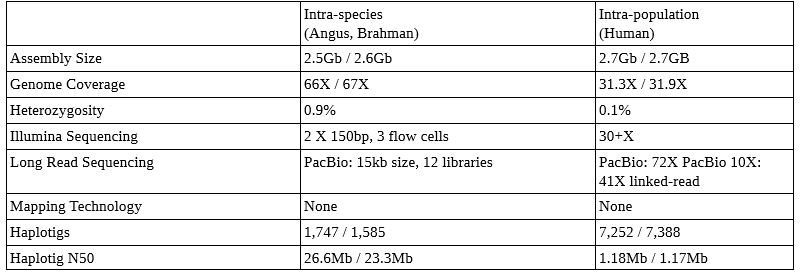
This technique can be applied within species (cow and yak with >1.2% heterozygosity– not published yet, but abstract is here), between species (Angus and Brahman with heterozygosity of 0.9%, above), and even humans (with heterozygosity of 0.1%, above). The canu software now ships with the ability to do trio binning (details on implementation here)! More detail in this presentation here.
GC content
Since we mentioned the difficulties in PacBio, we should also mention that Illumina has a genome structure sensitivity to watch out for as well, namely extreme GC content. Illumina sequencing will have lower coverage for very high or very low GC regions. This bias is important to consider, as it may require more sequencing to gain enough coverage of high and low GC regions if your organism has abnormal levels. Alternatively, relying more heavily on another technology (such as PacBio) might be beneficial.
Analysis Considerations
Depending how you plan on using your genome, you may want to tweak the general genome sequencing formula. For instance, population biology studies need a genome that is well assembled with high accuracy. Genomes help provide reliable SNPs, reduce genotyping error, and detect variations that are associated with genes—but only if those variations are not the result of technical errors in sequencing! Developing an erroneous SNP array can cost you—and the community—a lot more money than making sure you have addressed errors (this may have happened in Daphnia, and it affected the downstream microarray studies, as seen here).
Relying on higher accuracy technologies or increasing coverage is recommended to ensure proper assembly. Aiming for cheaper, lower-throughput technologies may allow you to have high coverage of the target individual while still preserving money for lower coverage of the rest of the population for variant analysis.
In other cases, annotation could be highly critical as well—such as in biomedical applications. A very non-model example of this is the Northern White Rhino. Building on the 2016 paper on induced pluripotent stem cells (iPSCs) from skin fibroblasts (paper here), researchers were able to produce iPSCs from Northern White Rhinos in a bid to save the species (described here). However, in order to confirm the iPSCs, they had to be able to identify markers for pluripotency through transcription similarity to stem cells in a very novel genome (abstract here). Additionally, they also had to pay particular attention to methylation signals of certain genes to develop plans for producing full sperm cells and eventually artificial insemination. The genome was critical, but the contiguity of the genome was less critical than the annotation in these analyses.
Northern White Rhino Genome

A male Northern White Rhinoceros. Photo: public domain
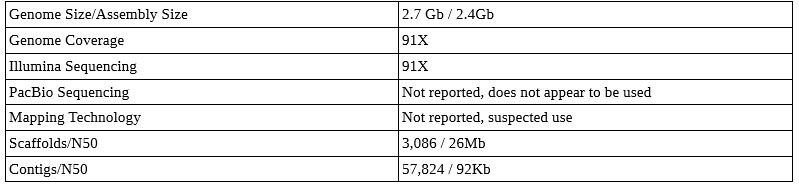
This genome paper is not published, but sequences are publicly available. This project also appears to have skipped PacBio sequencing, and it is unknown if mapping technology was used. I suspect it was – it is hard to get a good genome out of Illumina alone. Either way, note the much higher Illumina coverage needed to get good scaffold sizes.
Cost
While the sequence for a genome might be increasingly cheap to generate, the unmentioned costs are still high. Don’t forget computational and personnel time, and visualization! You may hear talk of sequencing a genome for ~$10k. This cost does NOT include person time to do the analyses, which is extensive. This cost does not include computational time, which can be expensive if you don’t plan accordingly.
Many of the papers linked in this blog refer to dozens of software packages for any one genome assembly. There were 23 different software packages just to assemble the genome/transcriptome for the Tegu paper. That number jumps to more than 44 packages needed when you include downstream analysis such as annotation. And that was for a genome without any major quirks.
Planning for analysis budget as well as a detailed analysis workflow from the get go will help keep these costs from ballooning. Running tests on smaller, publicly-available data sets will help you get familiar with the biases and complications of the software before investing in sequencing that may not be sufficient. Proper planning of the software and resources needed will save you from having to pay a grad student to figure out six different software packages that are all dead ends!
NOTE: This is the part we at NCGAS strive to help with: training people to plan and do these analyses, providing software support to save you from tearing your hair out trying to install dozens of packages, and providing or pointing you toward free-to-use machines capable of completing the demanding analyses. Contact us via our website here.
Alternatives

Bamboos sequenced by the GABR consortium. Source: doi:10.1093/gigascience/gix046
If sequencing technologies, budget, or personnel aren’t sufficient for doing your genome, it may be worth waiting a few years to approach a full genome. But this time does not need to be wasted. You may not need a genome right away to answer your questions. RNAseq and RADseq are two common stop-gaps until genomes are feasible.
RNAseq will get you a transcriptome for relatively little money and personnel time, and a lot of resources exist for these analyses (GigaScience has published a number of tools and toolkits). Most genome annotation requires a transcriptome anyway, so this step can both lead to a lot of useful research on the transcribed portions of the genome, while adding to future efforts. NCGAS has workflows (found here) and training materials (found here) for these analyses.
RADseq is a reduced library method that allows for the sequencing of parts of the genome. This data can be added to the sequence data generated for future genome analyses, but more often it allows for population biology studies to proceed without a full genome project completed. More information on the use of RADseq is available here. NCGAS has experience with this analysis as well, and is happy to help!

Sheri Sanders is a bioinformatician at the National Center for Genome Analysis Support (NCGAS).
NCGAS is a collaborative project between Indiana University and the Pittsburgh Supercomputing Center at Carnegie Mellon University. Their mission is “to enable the biological research community of the US to analyze, understand, and make use of the vast amount of genomic information now available.”
Further Reading:
Roscito JG, Sameith K, Pippel M, Francoijs KJ, Winkler S, Dahl A, Papoutsoglou G, Myers G, Hiller M. The genome of the tegu lizard Salvator merianae: combining Illumina, PacBio, and optical mapping data to generate a highly contiguous assembly. Gigascience. 2018 Dec 1;7(12). doi: 10.1093/gigascience/giy141.