Guest Blog: Data in the time of Coronavirus
 With much of the GigaScience team spanning the Hong Kong-Shenzhen border and now confined to remote working, the current 2019-novel coronavirus outbreak has been particularly disruptive and close to home. As with previous WHO “public-health emergency of international concern” such as Ebola and Zika, data has provided a potent tool in fighting both the outbreak, and the conspiracy theories that have filled the information gaps caused by poor communication and lack of trust in local governments. Compared to previous outbreaks, we are potentially better equipped to more rapidly fill in these information gaps. With real-time visualization of cases and results using tools like healthmaps, JHU Global Case dashboard, forkable method sharing via protocols.io, and Nextstrain, forums like virological. And the rise of preprints for sharing of results, bioRxiv & medRxiv having 48 Coronavirus submissions at time of posting (and causing some controversy with unreliable submissions, but seeing them debunked and retracted much faster than traditional peer reviewed publications).
With much of the GigaScience team spanning the Hong Kong-Shenzhen border and now confined to remote working, the current 2019-novel coronavirus outbreak has been particularly disruptive and close to home. As with previous WHO “public-health emergency of international concern” such as Ebola and Zika, data has provided a potent tool in fighting both the outbreak, and the conspiracy theories that have filled the information gaps caused by poor communication and lack of trust in local governments. Compared to previous outbreaks, we are potentially better equipped to more rapidly fill in these information gaps. With real-time visualization of cases and results using tools like healthmaps, JHU Global Case dashboard, forkable method sharing via protocols.io, and Nextstrain, forums like virological. And the rise of preprints for sharing of results, bioRxiv & medRxiv having 48 Coronavirus submissions at time of posting (and causing some controversy with unreliable submissions, but seeing them debunked and retracted much faster than traditional peer reviewed publications).
 Trying to digest some of these data streams and following previous blogs on the Ebola and Zika epidemics, we present another data oriented guest post from Michael Dean who has pooled together these various data streams to present a non-specialist view of the Coronavirus crisis. Michael is a researcher in the areas of human genetics and cancer. He has worked on the understanding of the role of host genetics in the response to HIV as well as cervical cancer and HPV. [Note: the material presented here is from Michael’s own perspective on the public Coronavirus data and does not represent the viewpoint of his employer].
Trying to digest some of these data streams and following previous blogs on the Ebola and Zika epidemics, we present another data oriented guest post from Michael Dean who has pooled together these various data streams to present a non-specialist view of the Coronavirus crisis. Michael is a researcher in the areas of human genetics and cancer. He has worked on the understanding of the role of host genetics in the response to HIV as well as cervical cancer and HPV. [Note: the material presented here is from Michael’s own perspective on the public Coronavirus data and does not represent the viewpoint of his employer].
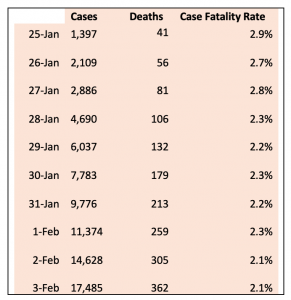
Table 1. Reported cases and deaths from 2019-nCoV. Source JHU CSSE
At the start of the Year of the Mouse, the world is experiencing an outbreak of a new Coronavirus, for now, called 2019-nCoV. As of February 4th (source JHU CSSE dashboard), there are 20,679 confirmed cases and 427 fatalities. Nearly 99% of the cases have been reported in China mostly in Hubei Province; and all except two reported fatalities have occurred in Mainland China. All the initial cases came from the city of Wuhan, the capital of Hubei, in Central China, but the origin of the virus is unknown. The case fatality rate (fatalities/total cases, CFR), based on reported data has been declining from nearly 3% to 2% as more data has become available, but is less than 2% in China, outside of Hubei. This is considerably lower than SARS (9.6%)1 and MERS (9-36%)2
The first full-length reference genome for the virus was deposited in GENBANK as NC_045512 by scientists at Fudan University, Shanghai, China, and referred to as the Wuhan seafood market pneumonia virus, as many of the initial cases were linked to a market of seafood and animals in the city of Wuhan. Multiple additional complete genomes have been deposited in GENBANK by the researchers within China and internationally including the US Centers for Disease Control (CDC). Additional isolates (62 in total at the time of posting) have been deposited in GISAID, a public repository of influenza virus sequences . Variation and evolution of the virus are being followed by an open-sourced project tracking pathogen evolution, Nextstrain. To date, the virus has shown few variable sites and low diversity.
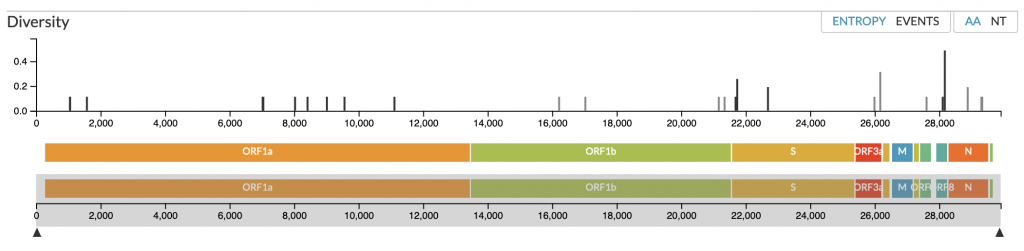
Fig 1. Map and diversity of 2019-nCoV. The map of the viral genome is shown with sites of identified variation and the known open reading frames (ORF) and viral proteins; spike (S), nucleocapsid phosphoprotein (N), and membrane glycoprotein (M). Source: Nextstrain.
The 2019-nCoV is most closely related to a beta-coronavirus isolated in bats in Yunnan3,4 (MG772933), and all the current isolates found in China or other countries are very closely related (Figure 2). The sequence data suggests that there was a single zoonotic event of viral transmission into the human population. Phylogenetic analysis places 2019-nCoV in the sarbecovirus subgenus that includes the SARS virus.3 Three prereprints in bioRxiv describe data indicating that the virus uses the angiotensin-converting enzyme II (ACE2) protein as its cellular receptor5-7, as does SARS.8 This result is significant as the Middle East respiratory syndrome (MERS) virus, MERS-CoV, uses dipeptyl peptidase 4 (DPP4; also known as CD26) as its receptor.9 To date, there is no variation in the portion of the virus that binds to ACE26.
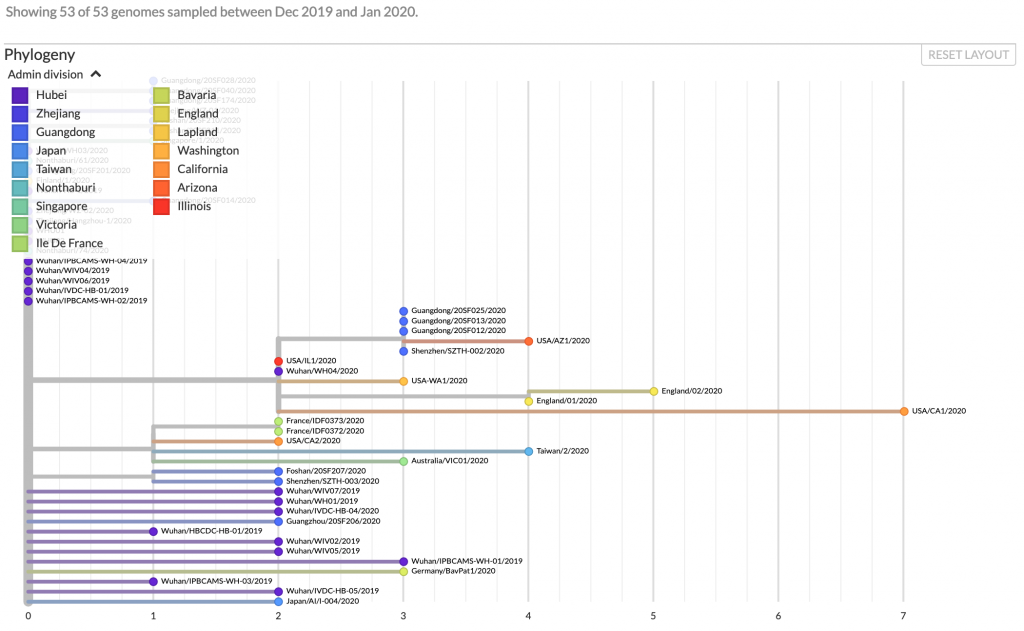
Fig 2. Phylogenetic tree of 2019-nCoV. A tree of sequenced isolates is shown along with the country or city where they were isolated, and the date. Source: Nextstrain.
Peer-reviewed publications documented the clinical features of 41 infected cases, 6 of whom died, and their demographic and clinical features.10 An estimate of 7 days from onset of symptoms to hospital admission was presented. The second description of 99 cases has also been published11 along with reports documenting human-human transmission.3,4 A third study of 425 cases provided an estimate of the mean incubation time of 5.2 days and a basic reproductive number of 2.2 (95% CI, 1.4 to 3.9).11 At the time of writing, current estimates of the case fatality rate (CFR) for 2019-nCoV are between 2 and 3 (Table 1). However, in regions of China outside of Hubei only 11 out of 6125 have died (0.18%). The first case report of a 35-year old male in the US described mild to moderate symptoms, and a virus with one one amino acid difference in one protein from the reference.12 There has been considerable cooperation among international health organizations, with the CDC publishing the details of a real-time PCR diagnostic test, and the World Health Organization has a site for up-to-date clinical treatment guidelines.
In conclusion there is still rapid growth of 2019-nCoV infections in Hubei Province, with a slowly declining fatality rate, but much fewer deaths outside of this region. Genetic and biological data is being rapidly produced and disseminated allowing for the rapid development of diagnostic tests and development of vaccines and other therapeutics.
We at GigaScience applaud this rapid and open sharing of the Coronavirus data. We already have stringent open and transparent data, review and publication policies in place, and would encourage submission of Data Notes and Technical Notes (methodological and software papers) to enable due credit to producers of data and tools. As with our dissemination of the data from the deadly German 2011 E. coli outbreak, curation and dissemination will be rapid, and we will also offer waivers of our article and data processing charges for Coronavirus studies to encourage this. Please contact us if you have presubmission inquiries or questions. While public events in our base of Hong Kong have all been called off, there are local “virtual” efforts to crowdsource and hack data from the outbreak, and some of the GigaScience team will be participating in a live streaming data science workshop on Sunday 9th February (follow the youtube link to watch).
Further Reading
1. de Wit . et al., SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14, 523-34 (2016).
2. Willman M et al., A Comparative Analysis of Factors Influencing Two Outbreaks of Middle Eastern Respiratory Syndrome (MERS) in Saudi Arabia and South Korea. Viruses 11(2019).
3. Zhu N. et al., A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med (2020).
4. Chan JF. et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet (2020).
5. Zhou P et al., Discovery of a novel coronavirus associated with the recent pneumonia outbreak in humans and its potential bat origin. bioRxiv (2020).
6. Hoffmann M et al., The novel coronavirus 2019 (2019-nCoV) uses the SARS-1 coronavirus receptor 2 ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv (2020).
7. Letko M et al., Functional assessment of cell entry and receptor usage for lineage B β-coronaviruses, including 2019-nCoV. bioRxiv (2020).
8. Li W et al., Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450-4 (2003).